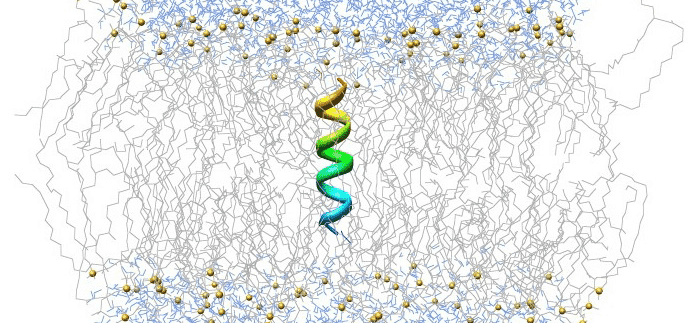
Running an MD Simulation of Graphene on Rescale
With the recent hype about graphene – a new material that has been praised for its unparalleled strength, lightness and electronic properties – I became curious and wondered whether I could run a molecular dynamics (MD) simulation of graphene given its simple chemical structure (hexagonal lattice). As someone new to the field of MD, with a background in front-end development, I cautiously set out to teach myself a thing or two about an MD simulation over a weekend and run a basic simulation on Rescale’s platform.
Molecular Dynamics
In layman’s terms, MD is a computer simulation about interactions between atoms (or molecules), using some basic laws of physics. More specifically, it is a method to determine the trajectories of atoms in phase space according to Newton’s equation of motion (F=ma); it does not try to solve quantum equation, however. In order to solve the equations of motion using a computer, it would take a person a long time before he/she masters the numerical methods required for basic math (e.g., differentiation, integration, etc). Fortunately, there are open-source tools — namely analysis codes — that provide optimized algorithms. So a person can focus his/her attention on solving physical questions.
Analysis Code
There is more than one analysis code suited for running an MD simulation. The list of codes I’ve found includes, CHARMM, AMBER, NAMD, GROMACS, DL_POLY, and LAMMPS. Among these six codes, the first four (CHARMM, AMBER, NAMD, GROMACS) are oriented toward biology (e.g. DNA force fields), while the last two are well suited for materials sciences, — the type I was after. In the end, I decided to use LAMMPS, mostly owing to its active community group and a well-written documentation.
Installing LAMMPS
First, I had to install LAMMPS on my machine to understand how an input file works in LAMMPS. Contrary to my initial worry about having to set up a linux environment, thanks to Derek Thomas, I was able to install the code directly onto my OS X environment using only a few commands, and was ready to run an MD simulation within minutes. For a simple sanity check, I followed the basic tutorial and ran the “in.obstacles” code (sample tutorial) using the following command:
lammps -in in.obstacles
And just to make sure that everything worked as intended, I ran the same job on Rescale, which yielded the same results as the one from my Macbook.
Now, a bigger challenge for me was to write a code that would alter conditions (i.e., temperature) and observe differences in carbon atoms’ physical properties (e.g., positions, kinetics energy, potential energy, etc) over time. However, setting up all this without making any mistakes in physics seemed difficult, so I decided to borrow an existing code from an academic paper.
Uniaxial Tensile Test in LAMMPS
After hours of googling, I found the paper “Molecular Dynamics Study of Effects of Geometric Defects on the Mechanical Properties of Graphene” by Nuwan Dewapriya Mallika Arachchige, which used LAMMPS to achieve a goal similar to what I was trying to do. The author was kind enough to attach the setup part of his experiment in the appendix, but I had to create my own sheet of graphene by writing a python script that determines the experiment boundaries and positions of carbon atoms.
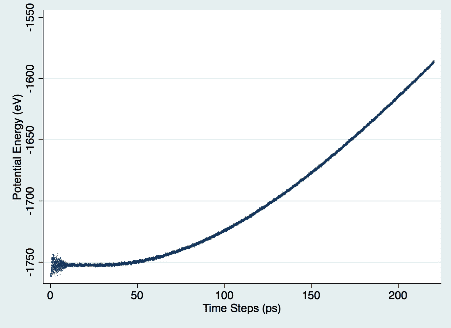
After trial and error, I confirmed that things worked correctly by comparing the rate of increase in potential energy that I found to the results in Archchige’s paper. Figure 1 demonstrates the potential energy increase over time.
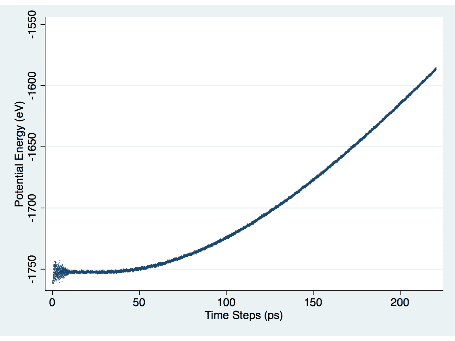
Figure 1. Potential Energy vs. Time
Rescale Platform
Having run a successful simulation on my machine, I decided to move it to the Rescale Platform for better performance. The size of my simulation was 166,320,000 (1.66e8) — the size of your experiment is determined by the product of the number of atoms (252) and the number of time-steps (660,000) in MD.
I ran the job on different hardware settings to demonstrate the performance gains for different core types. Figure 2 shows the differences in hardware performance for my simulation.

Figure 2. Hardware Performance Overview
Discussion
Overall, it was a fun first-time experience with an MD simulation. While graphing out the increase in potential energy over time was interesting, I see some room for creativity in terms of designing the experiment (e.g. people seem to be creating imperfect graphene models to see how a missing atom affects the stability of the overall structure).
As far as the computing power goes, by using Rescale’s Platform I was able to cut down my simulation time in a linear fashion. For those of you interested in running an MD simulation of Graphene, you can see the results of the simulation and download the input files here.
For information on how to set up and run LAMMPS simulations on Rescale, please visit rescale.com/resources/lammps.