並列化による Rescale で Gromacs をより高速に実行
グロマック ScalaLife によって開発され、欧州研究評議会によって資金提供されました。 Gromacs は、生化学分子の分子動力学を分析するために設計されたオープンソースの多用途パッケージです。 人気があるため、Gromacs を使用して、複数の dyno (つまり、コンピューティング コア) 間で単一のジョブを並列化するために必要な手順をデモンストレーションすることにしました。 オリジナルの基本的なGromacsチュートリアルを見つけることができます ここ
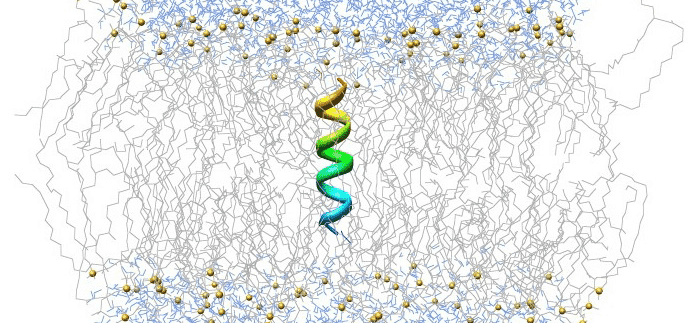
この例では、脂質ごとに 1024 個の水分子、合計 23 個の原子を含む二重層構成の 121,846 個のジパルミトイルホスファチジルコリン (DPPC) 脂質で構成されるリン脂質膜をシミュレートします。 大規模な問題が発生した場合は、実行時間を短縮するために複数の Dyno 間でジョブを並列化することを検討してください。 50 つの Dyno 間でジョブを正常に並列化できれば、ジョブの実行時間を XNUMX% 短縮できる可能性があります。
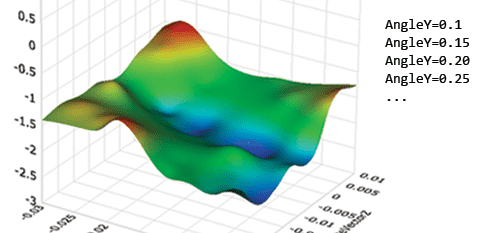
この例を XNUMX つの Dyno で並列化すると、ランタイムは半分に短縮されました。

ご覧のとおり、4 つのコアで並列化しても (XNUMX つのコアで実行した場合と比較した場合) ランタイムが XNUMX 倍向上することはありませんでした。 (a) ジョブの分解と (b) ジョブに関与するさまざまな Dyno 間のメッセージの受け渡しに関連して遅延が発生します。 さらに、分解が適切に行われない場合、結果として負荷の不均衡が生じ、さらなる遅延が発生します。
幸いなことに、Gromacs はユーザーにこれらすべての要素を追跡するための時間管理ツールを提供します。 これを使用するには、テキスト エディタで「job-name」.log ファイルを開くだけです。
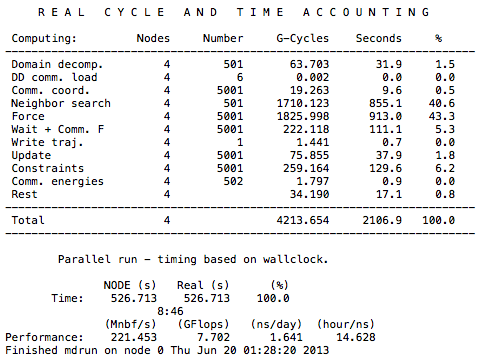
ここでの時間単位は CPU 秒です。 この値を関係する dyno の数 (この場合は XNUMX 台の dyno) で割ることで、ジョブの各部分に費やされる実時間を計算できます。 ドメインの分解、待機、通信、読み取り/書き込みは、理想的なスケーリングの達成を妨げる要因の一部です。 ただし、上級ユーザーは Gromacs が提供するデータを使用して、非効率な実行から学習し、より効率的な並列化を設計できます。
Rescale の Gromacs について詳しく知りたい場合は、こちらまでお問い合わせください。