Rescale でグラフェンの MD シミュレーションを実行する
グラフェン(比類のない強度、軽さ、電子的特性が称賛されている新素材)に関する最近の誇大宣伝を受けて、私は興味を持ち、その単純な化学構造 (六方格子) を考慮してグラフェンの分子動力学 (MD) シミュレーションを実行できないかと考えました。 )。 フロントエンド開発の背景を持つ MD の分野に不慣れな私は、週末に MD シミュレーションについて XNUMX ~ XNUMX つ独習し、Rescale プラットフォームで基本的なシミュレーションを実行することに慎重に着手しました。
分子動力学
平たく言えば、MD は、いくつかの基本的な物理法則を使用した、原子 (または分子) 間の相互作用に関するコンピューター シミュレーションです。 より具体的には、ニュートンの運動方程式 (F=ma) に従って位相空間内の原子の軌道を決定する方法です。 ただし、量子方程式を解こうとするわけではありません。 コンピュータを使って運動方程式を解くには、基礎的な数学に必要な数値的手法(微分、積分など)を習得するまでに長い時間がかかります。 幸いなことに、最適化されたアルゴリズムを提供するオープンソース ツール、つまり分析コードが存在します。 したがって、人は物理的な問題を解決することに注意を集中できます。
分析コード
MD シミュレーションの実行に適した解析コードは複数あります。 私が見つけたコードのリストには、CHARMM、AMBER、NAMD、GROMACS、DL_POLY、LAMMPS が含まれています。 これら XNUMX つのコードのうち、最初の XNUMX つ (CHARMM、AMBER、NAMD、GROMACS) は生物学 (DNA 力場など) を対象としていますが、最後の XNUMX つは材料科学に適しています。これは私が求めていたタイプです。 最終的に、LAMMPS を使用することにしました。主な理由は、活発なコミュニティ グループとよく書かれたドキュメントのおかげです。
LAMMPSのインストール
まず、入力ファイルが LAMMPS でどのように機能するかを理解するために、自分のマシンに LAMMPS をインストールする必要がありました。 Linux 環境をセットアップする必要があるのではないかという最初の心配に反して、おかげで デレク・トーマス、いくつかのコマンドだけを使用して、コードを OS X 環境に直接インストールすることができ、数分以内に MD シミュレーションを実行する準備が整いました。 簡単な健全性チェックのために、基本チュートリアルに従い、次のコマンドを使用して「in.obstacles」コード (サンプル チュートリアル) を実行しました。
ランプ - 障害物の中
そして、すべてが意図したとおりに機能することを確認するために、Rescale で同じジョブを実行しました。その結果、MacBook で実行したものと同じ結果が得られました。
さて、私にとってのより大きな課題は、条件 (温度など) を変更し、炭素原子の物理的特性 (位置、運動エネルギー、位置エネルギーなど) の違いを経時的に観察するコードを書くことでした。 しかし、物理学を間違えずにこれらすべてを設定するのは難しそうだったので、学術論文から既存のコードを借用することにしました。
LAMMPS での一軸引張試験
何時間もグーグルで検索した後、Nuwan Dewapriya Mallika Arachchige による論文「グラフェンの機械的特性に対する幾何学的欠陥の影響の分子動力学研究」を見つけました。この論文では、LAMMPS を使用して、私がやろうとしていたことと同様の目標を達成しました。 著者は親切にも実験の設定部分を付録に添付してくれましたが、私は実験の境界と炭素原子の位置を決定する Python スクリプトを書いて独自のグラフェン シートを作成する必要がありました。
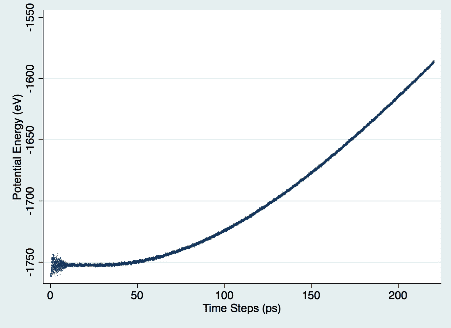
試行錯誤の末、私が見つけた位置エネルギーの増加率と Archchige の論文の結果を比較することで、正しく動作することを確認しました。 図 1 は、時間の経過に伴う位置エネルギーの増加を示しています。
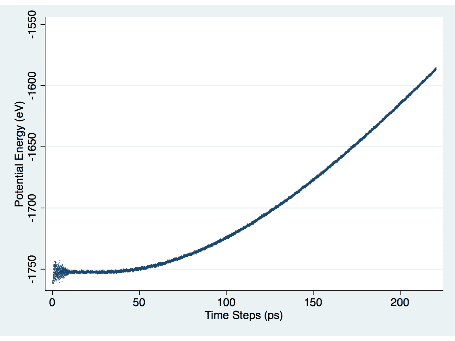
図 1. 位置エネルギー対時間
Rescale プラットホーム
私のマシンでシミュレーションを正常に実行した後、パフォーマンスを向上させるためにマシンを Rescale Platform に移行することにしました。 私のシミュレーションのサイズは 166,320,000 (1.66e8) でした。実験のサイズは、MD の原子数 (252) とタイムステップ数 (660,000) の積によって決まります。
さまざまなコア タイプでのパフォーマンスの向上を実証するために、さまざまなハードウェア設定でジョブを実行しました。 図 2 は、シミュレーションにおけるハードウェア パフォーマンスの違いを示しています。

図 2. ハードウェア パフォーマンスの概要
議論
全体として、初めての MD シミュレーション体験は楽しかったです。 時間の経過に伴う位置エネルギーの増加をグラフ化するのは興味深いことでしたが、実験の設計に関しては創造性の余地があると感じました (たとえば、欠落した原子が全体の構造の安定性にどのような影響を与えるかを確認するために、人々は不完全なグラフェン モデルを作成しているようです)。
コンピューティング能力に関して言えば、Rescale のプラットフォームを使用することで、シミュレーション時間を直線的に短縮することができました。 グラフェンの MD シミュレーションの実行に興味がある方は、シミュレーションの結果を確認し、入力ファイルをダウンロードできます。 こちら.
Rescale で LAMMPS シミュレーションをセットアップして実行する方法については、次のサイトを参照してください。 rescale.com/resources/lamps.