
カンザス大学生命科学研究

これは、Rescale の顧客である Eric Lee によるゲスト投稿で、彼の最初の Rescale 体験について説明しています。
こんにちは、みんな! 私の名前はエリック・リー、カンザス大学の大学院生です。 KU では、私の研究はナノ流体工学、特にナノウェッティングの問題に焦点を当てています。 ナノ流体工学は、ナノサイズの物体内およびその周囲のナノ流れの研究です。 ナノスケールでは、分散力、熱変動、流体力学的滑りなど、より大きな構造では観察されない物理的挙動が非常に重要になります。 ナノ流体工学は、PCR および関連技術用のラボ オン チップ デバイスの発明など、マイクロ流体デバイスをナノスケールまで小型化するための基礎です。
研究の一環として、コンピューター解析を実行するために LAMMPS などの分子動力学コードを使用します。 LAMMPS は、サンディア国立研究所によって配布されている古典的な分子動力学コードであり、 L年齢規模 Aトミック/Mオレキュラー M積極的に Pアラレル Sイミュレーター。 これは、単一のプロセッサ上で、またはメッセージ パッシング技術とシミュレーション ドメインの空間分解を使用して並列で実行されます。 コードは、新しい機能を追加して簡単に変更または拡張できるように設計されています。
LAMMPS のスケーラビリティを最大限に活用して、より多くの分析をより迅速に実行し、さまざまな結果を迅速に循環できるようにするには、LAMMPS とすぐに連携できる大規模なコンピューティング クラスターにアクセスする必要がありました。 Rescale については友人を通じて知り、実験に使ってみることを勧められました。
Rescale は、ユーザーがアップロードしたいカスタム スクリプトを含む、ドメイン全体で LAMMPS や他の多くのソフトウェア コードをサポートしていることに気付きました。 さらに、同社のハードウェアは最先端のもののようで、LAMMPS ジョブが Rescale でどれくらい高速に実行できるのか疑問に思いました。
Rescale がどのように機能するか、また、私の特定のバージョンの LAMMPS をサポートできるかどうかについて、最初はいくつかの質問がありました。 当初、Rescale には私の仕事に必要ないくつかのカスタム LAMMPS パッケージが含まれていませんでしたが、同社のエンジニアは非常に迅速にパッケージを実行できるようにしました。 私のすべての懸念事項に対する迅速な対応に感謝しました。Rescale サポートは通常、数時間以内に返事をくれました。 また、独自の LAMMPS ジョブをセットアップする方法を示す、非常に役立つスクリーンショットもいくつか送ってくれました。
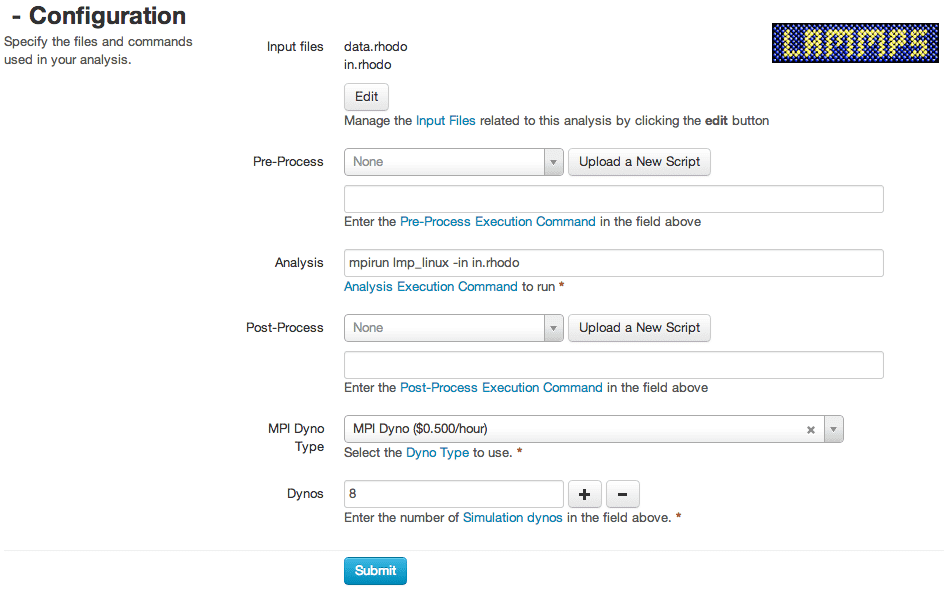
Rescale でジョブを実行するプロセスは非常に簡単です。 主な手順は次のとおりです。
(1) シミュレーションの名前を設定します。
(2) 推定されたシミュレーション ステップに基づいて、必要なコアの数を選択します (通常、コアが多いほど、シミュレーション時間は短くなります)。
(3) 入力ファイルと関連データファイルをアップロードする
(4) シリアル/パラレル解析タイプの選択
(5) ジョブを送信してください!
実行時のパフォーマンスに非常に感銘を受けました。 ローカル マシンで実行されている LAMMPS シミュレーションは、通常、完了するまでに約 4 日かかります。 Rescale では、同じ分析が 12 時間以内に終了しました。 この生産性の大幅な向上は、私の研究活動に大いに役立ちました。 これには本当に満足しており、Rescale でさらに多くのジョブを実行することを間違いなく楽しみにしています。 次回まで!
エリック・リーは大学院生です。 カンザス大学.