
University of Kansas, Life Sciences Research

This is a guest post by a Rescale customer, Eric Lee, discussing his first Rescale experience.
Hello everyone! My name is Eric Lee, and I am a graduate student at the University of Kansas. At KU, my research focuses on nanofluidics, specifically nanowetting problems. Nanofluidics is the study of nanoflows in and around nanosized objects. At the nanoscale, physical behaviors that are not observed in larger structures, such as dispersion forces, thermal fluctuations, and hydrodynamic slip, become very important. Nanofluidics is the basis for miniaturization of microfluidic devices down to the nanoscale, such as the invention of lab-on-a-chip devices for PCR and related techniques.
As part of my research, I use some molecular dynamics codes, such as LAMMPS, in order to run computational analyses. LAMMPS is a classical molecular dynamics code, distributed by Sandia National Laboratories and an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator. It runs on single processors or in parallel using message-passing techniques and a spatial-decomposition of the simulation domain. The code is designed to be easy to modify or extend with new functionality.
In order to take full advantage of the scalability of LAMMPS, so that I could run more analyses faster and cycle through various results quickly, I needed access to a large computing cluster that could work immediately with LAMMPS. I heard about Rescale through a friend, who recommended that I try using it for my experiments.
I noticed that Rescale supported LAMMPS and many other software codes across domains, including any custom scripts that users wanted to upload. In addition, their hardware appeared to be state-of-the-art, and I wondered how much faster my LAMMPS jobs could run on Rescale.
I had some initial questions about how Rescale worked, and whether it could support my specific version of LAMMPS. Initially, Rescale didn’t include some custom LAMMPS packages necessary for my jobs, but their engineers got the packages running very quickly. I appreciated their quick responses to all my concerns – Rescale support usually got back to me within a few hours. They also sent me some very useful screenshots showing how I could set up my own LAMMPS job.
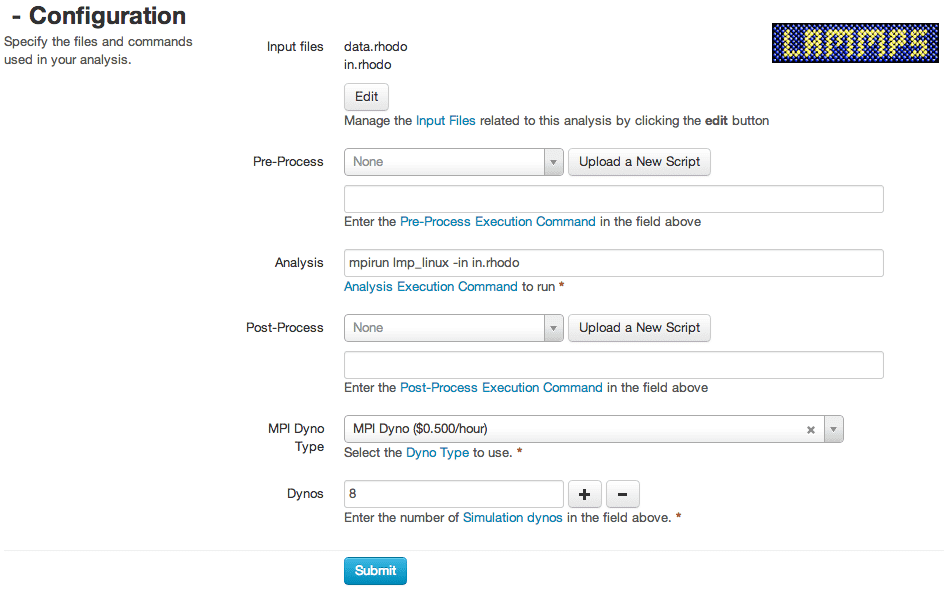
The process to run a job on Rescale is really simple. The main steps are:
(1) Set up a name for the simulation
(2) Choose number of cores needed based on estimated simulation steps (the more cores, the less simulation time usually)
(3) Upload the input file and associated data files
(4) Choose serial / parallel analysis type
(5) Submit the job!
I was very impressed with the runtime performance. My LAMMPS simulations running on my local machines usually take about 4 days to finish. On Rescale, the same analyses finished in less than 12 hours. This massive productivity gain helped my research efforts tremendously. I was really happy with this, and I am definitely looking forward to running more jobs on Rescale. Until next time!
Eric Lee is a graduate student at the University of Kansas.